首页
药品目录
白血病
血液科
肿瘤科
肝胆科
皮肤科
内分泌科
男科
妇科
肾内科
泌尿科
感染科
消化科
呼吸科
神经科
心脑科
眼科
精神科
免疫科
骨科
烧伤科
血管外科
疼痛科
移植科
内科
家族性高乳糜微粒血症综合征
白血病
缺铁
血栓性微血管病
ANCA相关性血管炎症
紫癜
真性红细胞增多症
华氏巨球蛋白血症
镰状细胞病
成人溶血
金属中毒
中粒细胞减少症
A型血友病
骨髓抑制
贫血
骨髓增生异常综合症(MDS)
血小板增多症
血小板减少症
骨髓纤维化
铁质积聚
止血
全部
免费咨询
白血病药物
Komzifti
用于治疗存在易感性核仁磷蛋白1(NPM1...
[ 详情 ]
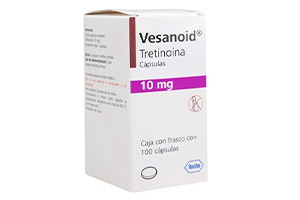
维甲酸胶囊
成人及≥1岁儿童的急性早幼粒细胞白血病(...
[ 详情 ]
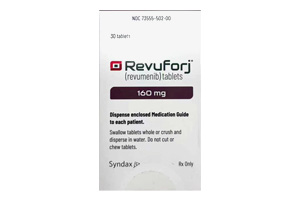
瑞维美尼
适用于1岁及以上KMT2A易位阳性复发/...
[ 详情 ]
艾代拉里斯
适用于复发性慢性淋巴细胞白血病(CLL)...
[ 详情 ]
他米巴罗汀
适用于经确诊为再发或难治性急性前骨髓球性...
[ 详情 ]
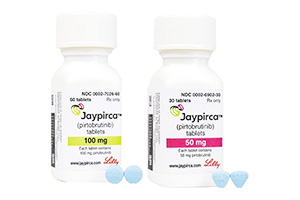
吡托布鲁替尼
适用于成年复发或难治性套细胞淋巴瘤患者(...
[ 详情 ]
奎扎替尼
适用于经FDA批准测试检测为FLT3-I...
[ 详情 ]
伊布替尼
适用于CLL/SLL、伴有17p缺失的 ...
[ 详情 ]
格拉斯吉布薄膜片
适用于与低剂量阿糖胞苷联合治疗新诊断的急...
[ 详情 ]
泽布替尼
适用于确诊为套细胞淋巴瘤、华氏巨球蛋白血...
[ 详情 ]
乌苯美司
确诊为成人急性非淋巴细胞白血病,且已完成...
[ 详情 ]
阿西米尼
存在费城染色体阳性慢性髓性白血病(Ph+...
[ 详情 ]
伐美妥司他
存在T细胞白血病淋巴瘤的成人患者。
[ 详情 ]
阿扎胞苷片
适用于成年急性髓系白血病患者,具体为在首...
[ 详情 ]
巯嘌呤片
适用于确诊为急性淋巴细胞白血病(ALL)...
[ 详情 ]
度维利塞
接受过至少两种全身治疗后复发或难治的慢性...
[ 详情 ]
艾德拉尼
适用于复发性慢性淋巴细胞白血病成年患者,...
[ 详情 ]
博舒替尼
确诊为慢性期、加速期或急变期Ph+CML...
[ 详情 ]
奈拉滨
1岁及1岁以上的T细胞急性淋巴细胞白血病...
[ 详情 ]
艾伏尼布
适用于经FDA批准的检测确认存在易感ID...
[ 详情 ]
上一页
1
/ 2
共34个
下一页
临床招募
PM8002联合紫杉醇对比化疗二线治疗小细胞肺癌
[适 应 症]
小细胞肺癌
[试验分期]
Ⅱ期
ES102联合特瑞普利单抗治疗晚期非小细胞肺癌
[适 应 症]
非小细胞肺癌
[试验分期]
Ⅱ期
比较Sigvotatug Vedotin与多西他赛治疗既往经治的非小细胞肺癌的研究
[适 应 症]
非小细胞肺癌
[试验分期]
Ⅲ期
QL2107或Keytruda®联合化疗治疗转移性非鳞状非小细胞肺癌的临床研究
[适 应 症]
非小细胞肺癌
[试验分期]
Ⅲ期
相关文章
相关信息
1
达沙替尼(Dasatinib)的适应症,用法用量,特殊人群用药
已帮助75人
2026-06-24 16:16:38
2
恩西地平的价格是多少钱一盒?
已帮助69人
2026-06-24 14:14:04
3
米哚妥林的价格是多少
已帮助160人
2026-06-18 15:13:07
4
米哚妥林怎么买?
已帮助161人
2026-06-18 14:54:41
5
恩西地平如何购买?
已帮助148人
2026-06-18 14:27:26
6
恩西地平多少钱一瓶?
已帮助236人
2026-06-18 14:11:01
7
Ziftomenib(Komzifti)怎么购买?
已帮助145人
2026-06-16 14:33:17
8
Ziftomenib国内哪里能买到?
已帮助144人
2026-06-16 14:27:26
9
恩西地平(Enasidenib)的适应症和不良反应
已帮助251人
2026-06-03 16:16:20
10
吉妥单抗(Mylotarg)的参考价格与疗效
已帮助187人
2026-06-03 16:09:11
吉妥珠单抗一旦使用了不能停吗?
吉妥珠单抗并不是一旦使用了不能停。如果用...
已帮助:531人
2026-06-25 14:29:30
吉妥珠单抗国内上市了吗?
截至2026年6月,吉妥珠单抗国内暂未正...
已帮助:529人
2026-06-25 14:31:07
Mylotarg中文名叫什么?
Mylotarg中文名叫吉妥单抗、吉妥珠...
已帮助:535人
2026-06-25 14:32:50
米哚妥林的用法用量
米哚妥林的剂量可因治疗的疾病不同而不同,...
已帮助:530人
2026-06-24 14:07:41
2026年米哚妥林纳入医保了吗?
截至2026年4月,米哚妥林(Midos...
已帮助:764人
2026-04-22 14:07:59
Midostaurin中文名叫什么?
Midostaurin中文名叫米哚妥林。...
已帮助:747人
2026-04-22 14:05:41
吉妥单抗是口服药还是注射药?
吉妥单抗(Mylotarg)是注射药。吉...
已帮助:769人
2026-04-15 13:55:51
米哚妥林(Midostaurin)是什么药?
米哚妥林(Midostaurin)是一款...
已帮助:775人
2026-04-13 14:05:18
恩西地平怎么吃?
恩西地平(Enasidenib)口服10...
已帮助:801人
2026-04-10 14:46:59
米哚妥林国内上市了吗?
截至2026年4月,米哚妥林(Midos...
已帮助:783人
2026-04-10 14:50:45
咨询热线:
400-001-2811
官方微博
官方客服
网站首页
药品目录
日本看病
临床招募
本网站不销售任何药品,只做药品信息资讯展示
药品医疗器械网络信息服务备案:(京)网药械信息备字(2025)第00185号
医伴旅
京公网安备 11011402012719号
京ICP备17022811号